Visualisation
The modbam output of of DNAscent detect and the bed files from DNAscent forkSense can be viewed directly in a genome browser. Here, we provide instructions of how to visualise BrdU and EdU calls together with fork and origin calls in IGV.
Download a recent version of IGV (https://igv.org/) that supports modified bases. After making sure the bam file from DNAscent detect is sorted and indexed, load this into IGV. You can change how BrdU and EdU calls are shaded by going to View –> Preferences –> Base Mods.
At the moment, BrdU and EdU are not directly supported by IGV (this is a planned update). In the meantime, IGV will parse EdU as 5fU and BrdU as 5caU. The colours of these bases can be changed independently. We suggest changing the base modification likelihood threshold to 0.5 to reduce clutter.

Right click on the bam track name and choose: Color alignments by –> base modification (all). You can then load any of the bed files produced by DNAscent forkSense for viewing alongside the BrdU and EdU calls.
The following image shows the result of a protocol where EdU (blue) was pulsed for five minutes followed by BrdU (red) for 10 minutes. We can observe the following replication origin from both the BrdU and EdU calls from DNAscent detect and the bed file from DNAscent forkSense.
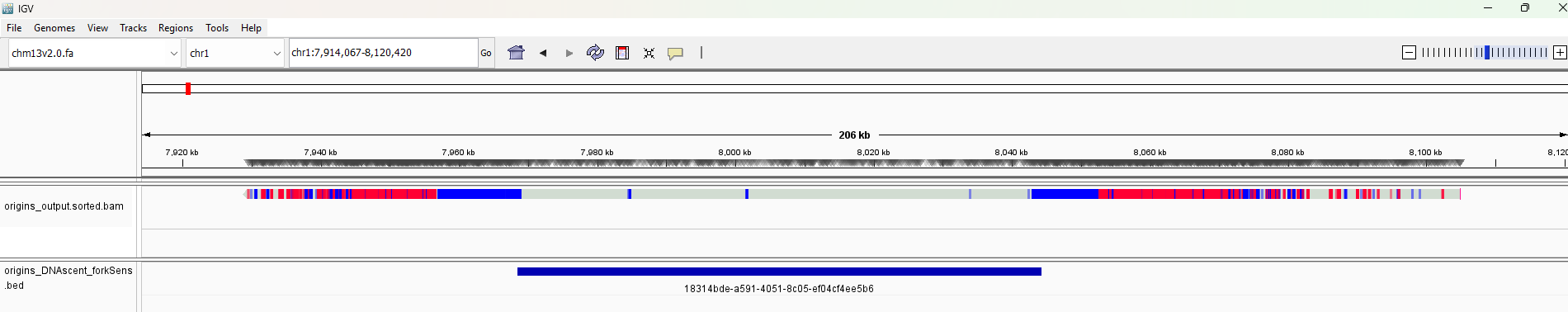
The bam files produced by DNAscent detect can, depending on your application, be large and unwieldy. If you have a bed file of origin calls from DNAscent forkSense called origins_DNAscent_forkSense.bed (or any other bed file from DNAscent forkSense) and a bam file from DNAscent detect called detect_output.bam, you can create a smaller bam file of just the reads with origin calls on them by doing the following:
awk '!/^#/ {print $4}' origins_DNAscent_forkSense.bed > origin_ids.txt
{ samtools view -H detect_output.bam; samtools view -h detect_output.bam | grep -F -f origin_ids.txt; } | samtools view -b - > origins.bam